[논문 정리] Conpair - 샘플간 swap 확인
- 생물정보학/논문 정리
- 2020. 8. 30.
1. Conpair
Tumor.bam과 Normal.bam을 Input으로 하여 동일한 개체에서 얻은 샘플인지 Concordance와 Contamination을 계산해주는 툴
* 해당 툴에 대해 다운로드 및 사용법정리 [Tools] - Conpair - tumor.bam normal.bam 비교
2. 과정
1) Pileup 생성
GATK Pileup을 이용해 bam파일로부터 pileup 파일을 생성.
Tumor.bam -> Tumor.pileup
Normal.bam -> Normal.pileup
2) Concordance 계산
marker의 MAF를 기반으로 Normal.pileup과 Tumor.pileup에서 bin별로 각각 아래의 확률 식으로 likelihood가 계산됩니다.
식 설명: k개의 read에 대해 j번쨰 base에서 A가 l번, B가 (k-l)번 나오는 것에대한 조건부 확률
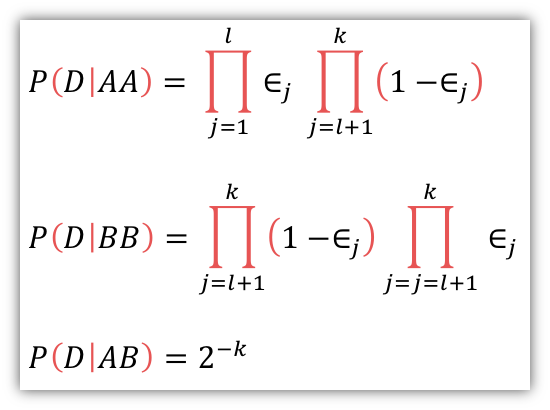
위 식의 계산 결과를 NL(Normal Likelihood)와 TL(Tumor Likelihood)에 딕셔너리 형태로 넣습니다. (키: AA, AB, BB, 값: 각각 조건부 확률값)
Markers(Bed 영역)을 한줄 씩 반복하며, m(bin)별로 NL과 TL의 likelihood의 최대값이 동일할 때가 concordance가 맞다고 보고 concordant에 1을 더해줍니다.
그렇지 않은경우 discordant에 1을 더해줍니다.

최종적으로 Concordance는 concordant / (concordant + discordant) 로 계산됩니다.
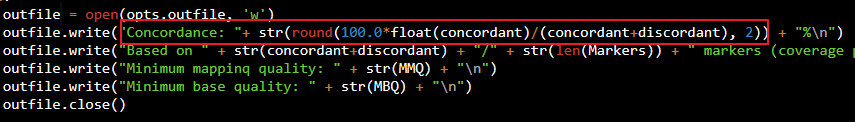
3) Contamination 계산
종양 샘플에서 발생하는 CopyNumber 변화는 이형 접합(heterozygous)에 대해 변화를 일으켜 오염 수준 추정하는데 어려움을 주기 때문에 동형 접합(homozygous)을 통해 종양샘플의 Contamination을 추정합니다.
순서
① 입력받은 Marker(bed 영역)에 대해 Normal 샘플에서 전체 bin에 대해 아래의 식을 통해 Contamination level(오염 수준)을 구합니다.

② Normal 샘플에 대해 Contamination level을 확인하면서, homozygous 영역에 대해 저장합니다.
③ Tumor 샘플에 대해서는 ②에서 저장한 homozygous 영역에 대해 위 통계식을 이용하여 Contamination을 계산합니다.
3. 한계
1) Marker 파일 생성 필요
논문에서 아래와 같이 Marker에 대해 MAF(4번째 칼럼)를 계산하였습니다.
-> 해당 파일을 생성해야 Concordance와 Contamination을 볼 수 있습니다.

2) Update 되지 않음
2018년 기준 더이상 업데이트가 되지 않아 기반 Tool들의 버전이 너무 낮습니다.
GATK 2.3버전을 사용했는데, 현재 구하기도 어려운 버전의 Tool 입니다.
해당 툴은 더이상 업데이트 되지 않기 때문에 해당 툴의 방법론만 학습하는것이 좋을것 같습니다.
'생물정보학 > 논문 정리' 카테고리의 다른 글
| [논문 정리] 8oxoG artifact (1) | 2020.09.05 |
|---|