Bam/Sam 파일 포맷: 인간유전체 정보를 담은 포맷
- 생물정보학/생물정보학 이론
- 2020. 8. 18.

안녕하세요
저번 포스팅에서는 DNA를 직접 NGS기기에 넣어 나오는 생(raw) 데이터 FASTQ에 대해서 알아보았습니다.
FASTQ : DNA 생(raw) 데이터!! 완벽 정리
FASTQ 데이터는 NGS기계에서 추출한 DNA 정보를 '데이터'형태로 보여주는 첫 파일입니다. 즉, FASTQ는 DNA 정보를 담고있는 아무 정제없는 가장 날 것의 데이터 입니다. 1. FASTQ의 구성 FASTQ는 아래 4줄이
bio-info.tistory.com
이번엔 이 FASTQ 데이터의 다음 단계인 Bam/Sam 파일이 뭔지, 어떻게 쓰이는건지 알아보겠습니다.
Bam 파일을 이해하려면 FASTQ 데이터를 알아야 합니다.
FASTQ 데이터는 read라고 불리는 매우 짧은 서열(50~200bp)들로 구성된 파일입니다. 보통 한 사람의 DNA를 NGS기기에 넣으면 개별 read의 개수는 백만 개가 넘으며, 경우에 따라선 1억 개가 넘기도 합니다. FASTQ는 DNA 조각들(read)의 염기서열 정보를 가지고 있지만, 어떤 염색체 어느 위치에 있는 DNA인지에 대한 정보는 가지고있지 않습니다.
목차
1. BAM 파일이란?
2. BAM 파일이 만들어지는 과정
3. BAM 파일 어떻게 쓸까?
1. BAM 파일이란?
보통 Bam파일은 FASTQ 파일로부터 만들어집니다. FASTQ파일이 짧은 DNA 서열(read)들이 무작위적으로 저장된 파일이라면, BAM파일은 DNA 서열을 쭉 이어 붙여 염색체 번호와 위치를 부여한 파일입니다. 예를 들어, FASTQ파일이 레고 부품이라면, Bam파일은 완성된 레고 로봇입니다.
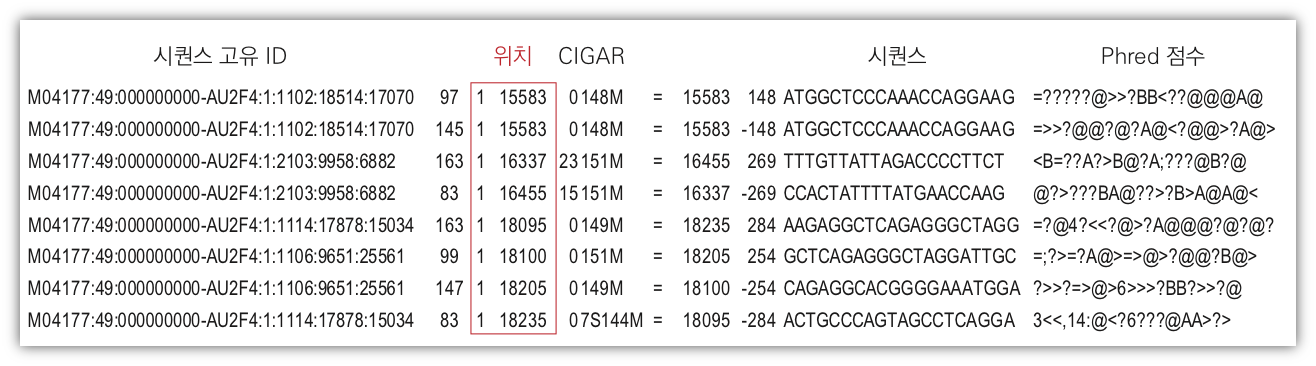
2. BAM 파일이 만들어지는 과정
1) 매핑(Mapping) / 정렬(Alignment)
FASTQ to SAM : FASTQ데이터에서 SAM 파일을 만들려면, 기존에 알려진 표준 유전체(reference genome)와 비교하여 어떤 염색체 어느 위치에 있는 건지 찾아주는 작업이 필요하며, 이를 매핑(mapping) 혹은 정렬(alignment)라고 합니다.
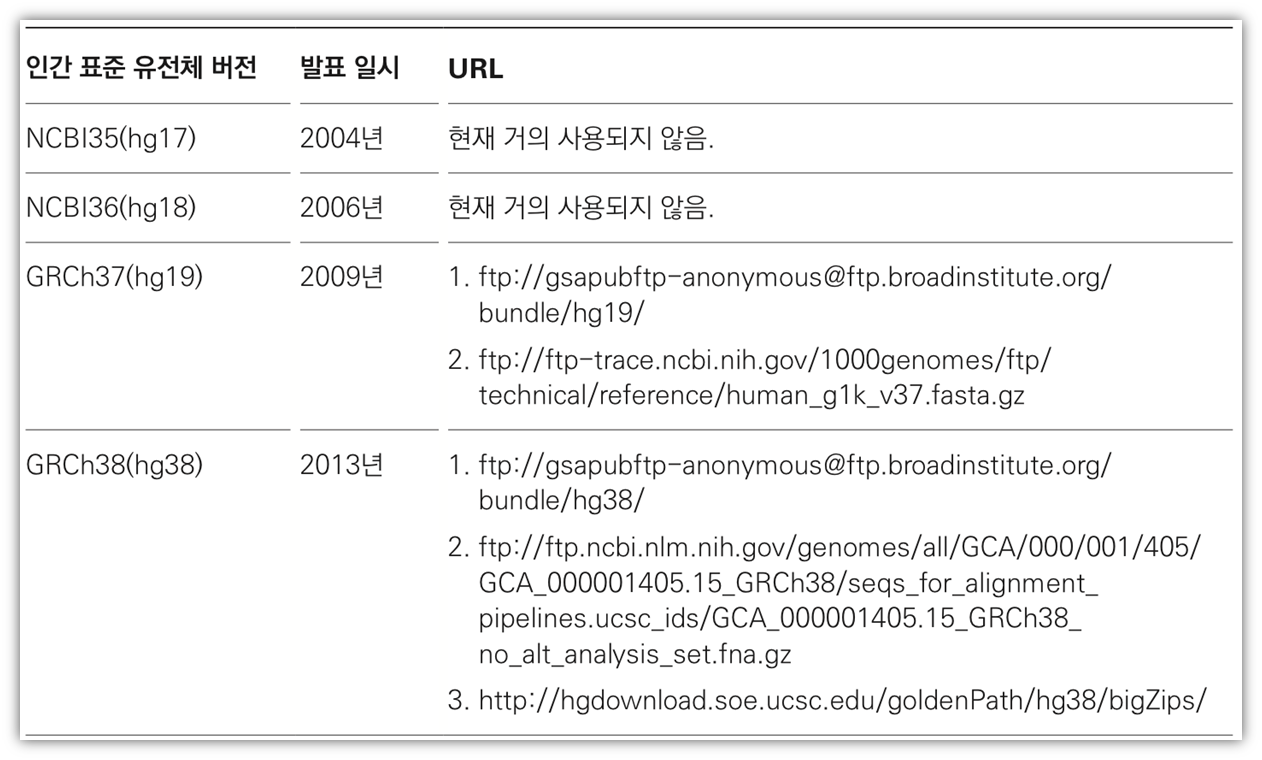
현재 가장 많이 쓰이는 인간 표준 유전체는 GRC(Genome Reference Consortium)에서 공개한 유전체 염기서열입니다. 크게 2가지 버전이 많이 쓰입니다.
첫 번째는 2009년 공개된 hg19(GRCH37) 버전입니다. 두 번째는, hg19 이후에 약점을 보완하고 개선된 hg38(GRCh38) 버전입니다. (2013년에 공개되었습니다. )
FASTQ파일의 Read들이 표준 유전체에 매핑이 완료되면 각 Read별로 표준 유전체 에서의 염색체 번호 및 위치가 기록됩니다. 이를 Sam 파일이라고 합니다.
이 과정은 BWA(Burrows-Wheeler Aligner)라는 알고리즘을 활용하여 진행됩니다. 이 알고리즘에는 BWA-ALN, BWA-SW, BWA_MEM 등의 서로 다른 버전이 있고, 가장 보편적으로 사용하는 알고리즘 버전은 BWA-MEM입니다. 뿐만 아니라, Novoalign이라는 BWA-MEM에 비해 분석 시간이 오래 걸리지만, 정확도는 더 우수하다고 알려진 알고리즘도 존재합니다.
2) Sam to Bam
Sam 파일은 ASCII(한글자당 7bit)로 구성되어 있습니다. 인간 유전체는 31억 개의 DNA 염기로 이루어져 있기 때문에 용량이 매우 큽니다. 그래서 용량을 줄이기 위해 이진수 형태의 Bam파일로 변환하여 사용합니다. (Sam파일과 Bam파일은 ASCII 코드로 되어있는지, 이진수로 되어있는지 차이만 있고 내용은 동일합니다.)
Sam파일을 Bam파일로 변환할 때는 보통 samtools를 사용합니다.
samtools view test.sam > test.bam
Bam파일은 Fastq파일로부터 왔기 때문에 Read들의 순서가 랜덤 합니다. 그래서 bam파일이 생성되면 보통 염색체와 위치 순(coordinate)으로 정렬해주고, 인덱스 파일을 생성해줍니다. 인덱스 파일을 생성하는 이유는 Bam파일이 정렬되었다 해도, 그 크기가 워낙 크기 때문에 인덱스로 접근하여 Bam파일 내에서 정보를 빠르게 찾기 위함입니다.
아래는 차례대로, 정렬해주고, 인덱스 파일을 생성하는 명령어입니다.
samtools sort test.bam
samtools index test.bam또한, samtools보다 비교적 빠르고 멀티스레드에 최적화된 툴인 sambamba를 이용하는 경우도 많습니다.

아래의 명령어를 보면 -o옵션(output)으로 test.bam을 만들고 sorting을 진행합니다. sambamba는 sort를 하면 인덱스 파일이 자동으로 생깁니다.
sambamba view test.sam -o test.bam
sambamba sort test.bam
3. Bam파일 어떻게 쓸까?
1) 시각화하여 돌연변이 검토
Bam파일이 완성되면 IGV(Integrative Genomics Viewer)를 통해 Read들이 정렬된 형태와 돌연변이가 어떻게 생겼는지를 시각화할 수 있습니다.
아래 그림은 KRAS 유전자가 있는 12번 염색체의 25,225,241위 치부터 25,226,497위 치 까지를 시각화 한 것입니다.
빨간색 막대기와 파란색 막대기가 있습니다. 이 막대기들이 Read입니다. 색깔이 다른 것은 방향이 다르기 때문입니다.
빨간색은 5'에서 3' 방향(5' to 3')이고, 파란색은 3'에서 5' 방향(3' to 5')입니다.
또한 아랫부분에 보면 파란색 Read에서 빨간 선이 하나 있는 부분이 보이는데, 저게 바로 돌연변이입니다.
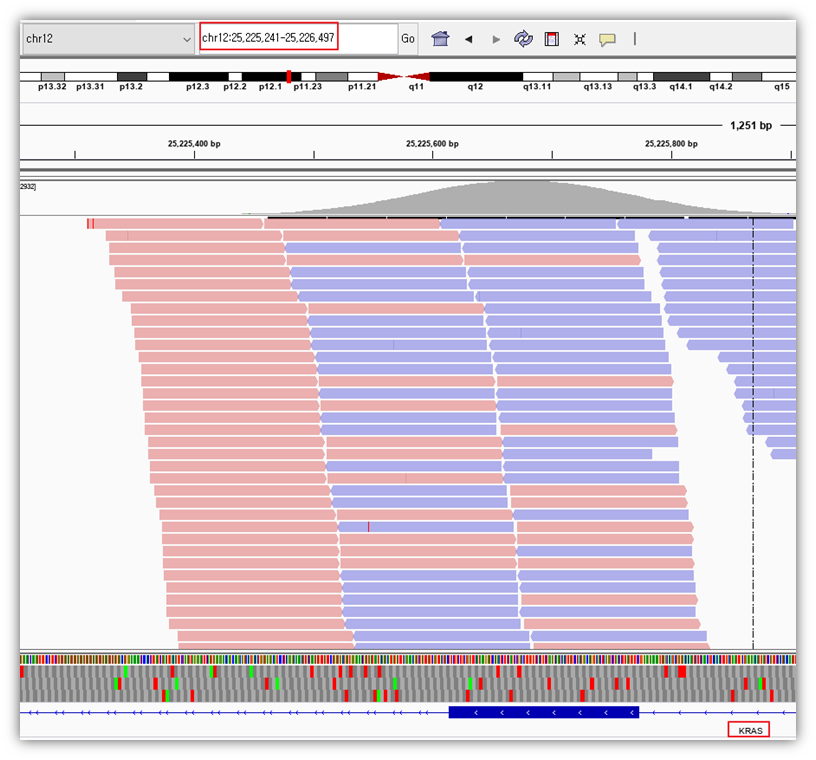
아래 사진에서 해당 돌연변이를 좀 더 확대하여 보겠습니다.
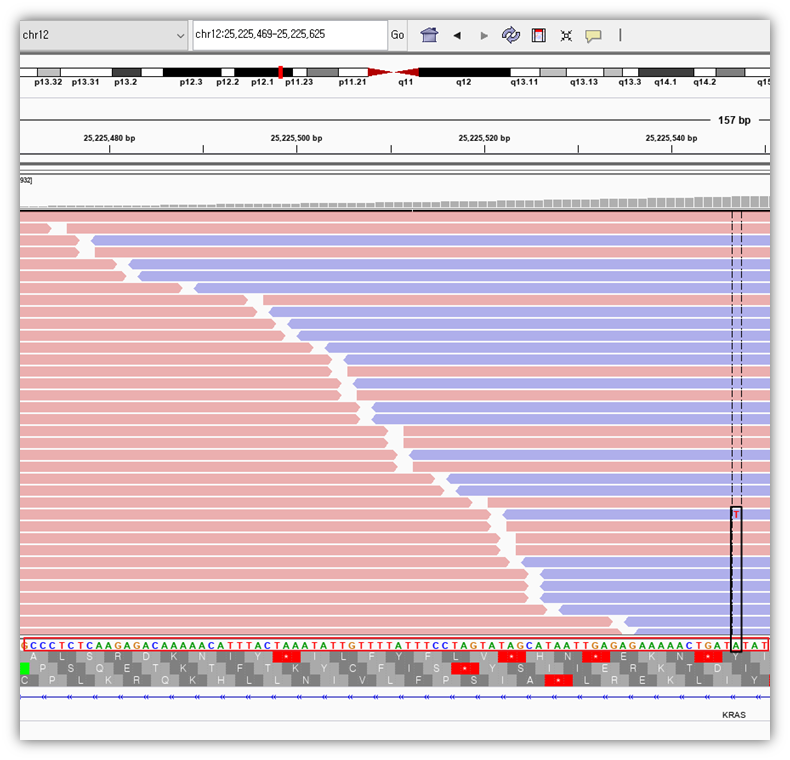
빨간색 선으로 돌연변이였던 부분이 빨간색 글씨로 T라고 써져있습니다. 그리고, 아래에 보면 색깔별로 알록달록하게 염기서열들이 표시되어있습니다. 아래에 있는 염기서열이 표준 유전체 서열입니다. 해당 부분은 표준 유전체 서열에선 A(아데닌)인데 해당 Read에선 T(티민)이어서 염기가 서로 다르기 때문에 돌연변이로 확인되었습니다.
해당 염기가 진짜 돌연변이 인지, 에러인지는 얼마만큼의 Read에서 신빙성 있게 나왔는지부터, 해당 변이의 Phred score까지 다양한 파라미터를 통해 확인하게 됩니다.
알록달록한 염기서열 아랫부분을 보면 염기 3개마다 알파벳 1개씩 써져있는 3줄이 보입니다. 해당 3줄은 아미노산 서열을 의미하며, Frame Shift가 일어날 수 있는 경우의 수가 총 3개 이기 때문에 3줄에 걸쳐 표현되었습니다.
2) 변이 검출 (Variant Calling)
보통 Bam 파일이 완성되면 어느 염색체 어느 부분에 돌연변이가 생겼는지 파악이 되었다고 볼 수 있습니다. 보통 DNA 데이터를 분석하는 목적은 질병과 돌연변이의 상관관계를 찾는 것입니다. 그런데 Bam파일은 Read들도 너무 많고, 정상 서열 정보도 너무 많습니다. 그래서 필요한 정보인 돌연변이 정보만 추출해서 사용하는 것이 효율적입니다. 그래서, 변이 정보만 추출하는 단계인 변이 검출(Variant Calling)을 Bam파일 이후에 진행하게 됩니다.
Variant Calling 단계는 다음 시간에 자세히 알아보겠습니다.
이번 시간에는 BAM파일이 뭔지, 어떻게 만들어지는지, 그리고 어떻게 쓰는지에 대해 자세히 알아보았습니다.
다음에 더 유익한 글로 찾아오겠습니다.
읽어주셔서 감사합니다.
Reference)
그림 1) 그림 2) : 이승태, 이경아, 심효섭 외 6명, NGS 기반 유전자 검사의 이해 (식품의약품 안전평가원), 23p - 35p
그림 3): http://lomereiter.github.io/sambamba/
'생물정보학 > 생물정보학 이론' 카테고리의 다른 글
| NGS - DNA 데이터 분석 한장 요약 (2) | 2020.08.26 |
|---|---|
| Rosalind란? 생물정보학 알고리즘 문제 풀기 (0) | 2020.08.19 |
| FASTQ 파일 포맷 : DNA 생(raw) 데이터 정보를 담은 포맷 (0) | 2020.08.04 |
| 타겟 선별과 NGS 시퀀싱 종류 (WGS, WES, Target-seq) (0) | 2020.07.28 |
| Central Dogma(중심원리)란? (4) | 2020.07.02 |